
Nickel-Chrom-Legierungsschichten lassen sich aus einem Chrom(III)elektrolyten auf Sulfatbasis mit Zusätzen (Komplexbildner, pH-Puffer, Leitsatz, organische Additive) abscheiden. Aufgrund der großen Differenz zwischen den Abscheidepotentialen von Nickel und Chrom hängt die Legierungszusammensetzung stark von der Stromdichte ab, weshalb die Nickelabscheidung durch Diffusionslimitierung begrenzt werden muss, um einen höheren Chromanteil in der Schicht zu erzielen. Bei hohen Stromdichten und niedriger Nickelkonzentration werden chromreiche Schichten mit bis zu 64 Gew.% Chrom abgeschieden. Durch Zugabe von Saccharin können der Chromgehalt sowie die Stromausbeute gesteigert werden. Die Schichten enthalten Kohlenstoff und bei Abscheidung aus einem Elektrolyten mit Saccharin geringe Mengen an Schwefel. Die größten Herausforderungen bei der Abscheidung von Nickel-Chrom-Schichten sind die hohen inneren Spannungen in den mit Gleichstrom abgeschiedenen Schichten, die zur Rissbildung führen. Bei Anwendung von Pulse-Plating ist es möglich, rissfreie Schichten mit Dicken von mehr als 10 μm abzuscheiden. Die Schichten wachsen kompakt mit einer leicht globularen Struktur auf.
1 Einführung
Hartchromschichten werden häufig zur Verbesserung der Verschleiß- und Korrosionsbeständigkeit von technischen Bauteilen eingesetzt. Zum gegenwärtigen Zeitpunkt werden für industrielle Anwendungen Elektrolyte auf Basis von sechswertigen Chromverbindungen verwendet. Aufgrund der von Chrom(VI)verbindungen ausgehenden Gesundheits- und Umweltgefahr wird deren Einsatz jedoch zunehmend reglementiert. Die Entwicklung einer alternativen Oberflächenbeschichtung als Ersatz für die Hartchrombeschichtung aus chrom(VI)basierten Elektrolyten kann daher als ein Schlüsselproblem der modernen Oberflächentechnik angesehen werden [1]. Eine Alternative stellt die Chrombeschichtung auf Basis eines Chrom(III)elektrolyten dar. Die Korrosions- und Verschleißbeständigkeit solcher Schichten ist jedoch geringer als die einer konventionellen Hartchromschicht aus einem chrom(VI)basierten Elektrolyten. Durch das Zulegieren weitere Elemente können die Schichteigenschaften weiter verbessert werden [2].
Großes Potential zeigen dabei Nickel-Chrom-Legierungsschichten. Für die galvanische Abscheidung von Nickel-Chrom-Legierungsschichten ist bisher kein kommerzielles Verfahren bekannt. Auch wenn die ersten Arbeiten zu diesem Themengebiet bereist einige Jahrzehnte zurückgehen [3], befindet sich deren Entwicklung noch am Anfang, was sich ferner an der Anzahl an wissenschaftlichen Publikationen und veröffentlichten Patenten zeigt. In der vorliegenden Arbeit wurden Untersuchungen zur galvanischen Abscheidung von Nickel-Chrom-Legierungsschichten aus einem sulfatbasierten Chrom(III)elektrolyten durchgeführt. Neben dem Einfluss von Stromdichte, Temperatur und der Nickelkonzentration im Elektrolyten wurde die Anwendung von Pulse Plating untersucht.
2 Stand der Technik
Chrisholm et al. geben in ihrer Arbeit eine Übersicht über die Anfänge der Entwicklung zur Abscheidung von Nickel-Chrom-Schichten [3]. Zu diesem Zeitpunkt lag der Fokus noch auf Elektrolyten auf Basis von sechswertigen Chromverbindungen. In neueren Veröffentlichungen werden jedoch ausschließlich dreiwertige Chromverbindungen eingesetzt.
Um die Abscheidegeschwindigkeit zu erhöhen, werden dem Elektrolyten Komplexbildner zugesetzt. Typische Komplexbildner sind Carbonsäuren wie beispielsweise Ameisensäure, Oxalsäure, Citronensäure, Äpfelsäure, Malonsäure, Glycolsäure oder Glycin und Harnstoff. Voraussetzung für die Reduktion des Metall-Aqua-Komplexes ist, dass die Metallionen an der Kathode die Elektronen aufnehmen können und dadurch reduziert werden. Die Chrom(III)ionen sind in Form eines regulären Oktaeders von sechs Wassermolekülen umgeben, wodurch sie die Kathode nicht berühren können. Vor der Abscheidung müssen die Wassermoleküle abgestreift werden, bezeichnet als Dehydratisierung.Die Dehydratisierung ist jedoch aufgrund des relativ kurzen Abstands und der starken Bindung zwischen den Chrom(III)ionen und den Wassermolekülen schwierig. Durch die Zugabe von geeigneten Komplexbildnern kann die Dehydratisierung allerdings beschleunigt werden [4].
Damit der Vorgang der Komplexbildung ablaufen kann, müssen die Komplexbildner thermodynamisch stabilere Komplexe als Wasser bilden. Die Stabilität des Komplexes sollte jedoch nicht zu hoch sein, da sonst die Geschwindigkeit der Chromabscheidung deutlich abnimmt. Neben der thermodynamischen Stabilität der gebildeten Komplexe ist die Kinetik der Komplexbildung von großer Bedeutung. Die Reduktion von Chrom ist ein mehrstufiger Prozess, der über die Zwischenstufe Chrom(II) abläuft. Das Chrom(II)ion ist in wässriger Lösung jedoch nicht stabil und wird leicht zu Chrom(III) oxidiert. Ein geeigneter Komplexbildner sollte in der Lage sein, das gebildete Chrom(II) durch eine Komplexierung gegen die Oxidation zu stabilisieren. Auf diese Weise wird ein hohes Angebot des als Zwischenstufe entstandenen Chrom(II) gewährleistet. Damit ist die Kombination aus den optimalen kinetischen Bedingungen für die Komplexierung von Chrom(II) und der thermodynamischen Stabilität der Chrom(III)ionen von primärer Wichtigkeit für den Gesamtprozess. Da thermodynamische und kinetische Daten für die Chromkomplexbildung nur selten vorhanden sind, ist die Auswahl eines geeigneten Komplexbildners oftmals nicht ohne weiteres möglich beziehungsweise mit einem höheren Aufwand verbunden [5].
Aufgrund der dargestellten Problematik schlägt Datta [5] die Verwendung von zwei Komplexbildnern vor. Neben Citronensäure wird dem Elektrolyten ein zweiter (nicht genannter) Komplexbildner zugesetzt, der mit Chrom(II) schnell Komplexe ausbilden soll. Der Komplexbildner wird auch als Katalysator beziehungsweise Regenerator bezeichnet, da er nach der Chromabscheidung wieder frei wird und zudem reduzierende Eigenschaften aufweist [5].
Bei Verwendung von Carbonsäuren als Komplexbildner werden meist größere Mengen an Kohlenstoff (als Abbauprodukt der Carbonsäure) in die Schicht eingelagert. Danilov et al. untersuchten die Kinetik und mögliche Mechanismen der galvanischen Abscheidung von Chrom-Kohlenstoff-Legierungen aus einem Elektrolyt auf Basis von Chrom(III)sulfat mit Harnstoff und Formiat als organische Komponente. Es wird angenommen, dass der Mechanismus des Kohlenstoffeinbaus eine chemische Wechselwirkung der adsorbierten organischen Moleküle mit hochaktiven Chrom-Ad-Atomen ist, was im Grunde einen nicht-Faraday-Prozess darstellt [6].
Als Metallsalz werden ausschließlich Sulfate oder Chloride eingesetzt. Der Grund liegt in der breiten und günstigen Verfügbarkeit. Vereinzelt werden für Elektrolyte auf Basis von dreiwertigen Chromverbindungen auch Methansulfonate oder Ammoniumchromalaun verwendet. Danilov et al. stellten fest, dass die Verwendung von basischem Chromsulfat gegenüber saurem Chromsulfat den Vorteil hat, dass die Metallabscheidung bereits bei einer geringeren Stromdichte einsetzt und die Deckfähigkeit des Elektrolyten erhöht wird. Begründet wird das durch Hydroxidionen, welche in der inneren Sphäre der Chromkomplexe vorhanden sind und eine beschleunigte Entladung von Chrom(II) fördern [7].
Die Reduktion von Chrom(III) zum Chrommetall (Chrom(0)) ist durch ein Standardreduktionspotential (-0,74 V) gekennzeichnet, das deutlich negativer ist, als das Standardreduktionspotential von Wasserstoff (0 V). Eine weitere Eigenschaft ist die sehr geringe Überspannung der Wasserstoffabscheidung auf Chrom und die sehr hohe Überspannung der Chromabscheidung auf Chrom. Die Metallabscheidung erfolgt daher mit einer beträchtlichen Wasserstoffentwicklung, welche einen signifikanten Anstieg des pH-Werts an der Elektrodenoberfläche verursacht. Bei hohen pH-Werten wiederum hydrolysiert der Komplex [Cr(H2O)6]3+ und bildet eine Reihe von Reaktionen, die als Endprodukte Polymere mit hohem Molekulargewicht enthalten; bei diesen sind die Chrom(III)ionen über Hydroxylbrücken verbunden. Dieses Phänomen wird als Olation bezeichnet. Die gebildeten Polymere fallen nahe der Kathodenoberfläche aus, was die Verfügbarkeit an Chrom(III) einschränkt und die Stromausbeute senkt [8]. Bei hohen pH-Werten kann es zudem zur Ausfällung von Metallhydroxiden kommen. Um dies zu verhindern, wird dem Elektrolyten als pH-Puffer meist Borsäure zugegeben. Borsäure ist eine Lewis-Säure, welche durch Aufnahme von Hydroxidionen sauer wirkt. Vereinzelt wird auch über die Verwendung von Aluminiumsulfat als Puffer berichtet [9]. Dabei handelt es sich um eine Brönsted-Säure, welche durch Abgabe von Protonen sauer wirkt.
Elektrolyte für die Abscheidung von Nickel-Chrom-Schichten weisen eine geringe elektrische Leitfähigkeit auf. Das führt zu einem hohen ohmschen Spannungsabfall im Elektrolyten, wodurch sich der Elektrolyt während der Abscheidung stark erwärmt. Zudem muss eine höhere Abscheidespannung angelegt werden, was in der praktischen Anwendung mit höheren Energiekosten verbunden ist. Um den Spannungsabfall möglichst gering zu halten, werden dem Elektrolyten hohe Konzentrationen an Leitsalzen zugegeben. Bevorzugt werden Natriumsulfat-/chlorid oder Ammoniumsulfat/-chlorid eingesetzt.
Neben Metallsalz, Komplexbildner, Leitsalz und pH-Puffer werden dem Elektrolyten Additive, meist auf organischer Basis, zugesetzt. Zur Verhinderung einer Oxidation von Chrom(III) zu Chrom(VI) an der Anode, werden oftmals Bromid- oder Fluorid-Verbindungen verwendet. Aufgrund der geringen Stromausbeute und der damit einhergehenden starken Wasserstoffentwicklung ist die Verwendung von Netzmitteln beziehungsweise Tensiden notwendig. Diese verringern die Oberflächenspannung des Elektrolyten, wodurch das Anhaften von Wasserstoffbläschen erschwert und die Ausbildung von Pittings (lochartigen Vertiefungen) in der Schicht verringert wird. Zudem verhindern sie den Austrag von Aerosolen aus dem Elektrolyten.
Langleis und Nepper untersuchten die Abscheidung von Nickel-Eisen-Chrom-Schichten aus einem Chrom(III)elektrolyten. Der von ihnen beschriebene Elektrolyt wurde abgeleitet von einem von Datta entwickelten Ansatz. Die Zugabe von 0,5 g/L Natriumdisulfit wurde als positiv für die Chromabscheidung ermittelt. Die Stromausbeute der Chromabscheidung konnte dadurch um den Faktor vier auf etwa 12 % erhöht werden. Ohne die Zugabe von Natriumdisulfit wurde überwiegend das edlere Eisen abgeschieden. Der Einsatz von Natriumdisulfit führte zudem zu einem Einbau von Schwefel in die Schicht. Als möglichen Mechanismus geben sie die von Datta und Hoare vorgeschlagene katalytische Wirkung von Hydrogensulfit an [10, 11].
Leimbach et al. befassten sich mit dem Einfluss von Saccharin (Benzoesäuresulfimid) auf die Abscheidung von dekorativen Chromschichten. Die Zugabe von 1,5 g/L Saccharin erleichterte die Reduktion von Chrom wodurch die Stromausbeute von 2 % auf 8 % gesteigert werden konnte. Als möglicher Mechanismus wird angenommen, dass Saccharin mit Chrom(II) einen Komplex bildet, wodurch dieses stabilisiert und die Reduktion von Chrom(II) zu Chrommetall entsprechend gefördert wird [12].
Als Anoden kommen ausschließlich inerte Anoden zum Einsatz. Aufgrund der geringen kathodischen Stromausbeute würde die Verwendung von löslichen Anoden eine starke Anreicherung der Chrom- beziehungsweise Nickelkonzentration verursachen. Bei Verwendung von unlöslichen Anoden ist darauf zu achten, dass das für die Sauerstoffentwicklung notwendige Potential an der Anode so weit abgesenkt werden muss, dass dieses unterhalb des Potentials für die Oxidation von Chrom(III) zu Chrom(VI) liegt und es ausschließlich zu einer Sauerstoffentwicklung kommt. Bohnet untersuchte verschiedene Anodensysteme. Das beste Ergebnis zeigten demzufolge Anoden mit Mischoxidbeschichtung, da bei diesen nahezu keine Oxidation des Chroms erfolgt [13]. Ein weiterer Vorteil von Mischoxidanoden ist, dass die oftmals als Komplexbildner verwendet Ameisensäure nicht oxidiert (und damit für die Abscheidereaktionen unwirksam wird). Platin hingegen zeigt eine starke spezifische Adsorption von Ameisensäure und weist eine hohe katalytische Aktivität gegenüber der Ameisensäureoxidation auf [14].
Den größten Einfluss auf die Legierungszusammensetzung nimmt die Stromdichte. Aufgrund des unterschiedlichen kathodischen Polarisationsverhaltens bei der Abscheidung von Nickel und Chrom aus einem Elektrolyten kann der Chromgehalt in der abgeschiedenen Schicht nahezu beliebig eingestellt werden. Der Chromgehalt in der hergestellten Nickel-Chrom-Schicht nimmt mit steigender Stromdichte zu. Durch Erhöhung der Stromdichte von 15 A/dm2 auf 30 A/dm2 konnten Huang et al. den Chromgehalt von 15 Gew.% auf 85 Gew.% steigern [15]. Bei zu hohen Stromdichten kommt es jedoch aufgrund der verstärkten Wasserstoffentwicklung zu einem starken Anstieg des pH-Werts im kathodennahen Raum und zu einer Ausfällung von Metallhydroxiden [12].
Die Beschichtungstemperatur wird meist zwischen 30 °C bis 40 °C angegeben. Mit steigender Temperatur erhöht sich die Diffusionsgeschwindigkeit der Metallionen und die kathodische Polarisation nimmt ab. Die Trends einer Erhöhung der Diffusionsgeschwindigkeit und einer Verringerung der Kathodenpolarisation mit zunehmender Temperatur sind für Chrom(III)ionen kleiner als für Nickel(II)ionen (in den hier betrachteten Elektrolyten), was den Chromgehalt in der Beschichtung verringert. Mit sinkender Temperatur des Elektrolyten nimmt auch die Gleichmäßigkeit der Beschichtung ab. [16].
Der Chromgehalt der Beschichtung nimmt mit zunehmendem pH-Wert des Elektrolyten zu. Je höher der pH-Wert ist, desto höher ist die Dissoziationsgeschwindigkeit des Komplexbildners. Daraus resultiert eine höhere Konzentration an Chrom(III)ionen an der Kathodenoberfläche (bzw. in Nähe der Kathodenoberfläche) und eine höhere Entladungsgeschwindigkeit. Chrom(III) nimmt jedoch leichter an der Polymerisationsreaktion teil, wenn der pH-Wert zu hoch ist. Es bildet sich ein Hydroxidfilm auf der Elektrodenoberfläche, welcher eine Entladung von Chrom(III)ionen verhindert [16].
Eine der größten Herausforderungen bei der Abscheidung von Nickel-Chrom-Legierungsschichten sind die hohen inneren Spannungen in den abgeschiedenen Schichten und die daraus resultierende Rissbildung. Die Abscheidung von größeren Schichtdicken, wie sie für technische Zwecke benötigt werden, ist dadurch erschwert [17]. Die hohen inneren Spannungen sind auf die Bildung von Chromhydriden zurückzuführen [18]. Aufgrund der geringen kathodischen Stromausbeute geht die Metallabscheidung mit einer starken Wasserstoffentwicklung einher. Aus kinetischen Gründen bildet sich zuerst ein Chromhydrid, welches nach kurzer Zeit zum thermodynamisch stabileren elementaren Chrom zerfällt. Dieser Schritt geht mit einer Volumenkontraktion einher, da Chromhydride ein hexagonales Gitter aufweisen und Chrom kubisch raumzentriert vorliegt. Die Volumenkontraktion führt zu hohen Zugspannungen in den abgeschiedenen Schichten, welche sich schließlich unter Rissbildung abbauen.
3 Experimenteller Teil
Anhand der Literaturrecherche und durchgeführten Vorversuchen wurde ein Elektrolytansatz abgeleitet (Tab. 1). Der Elektrolyt enthält neben den beiden Metallsalzen Nickelsulfat und basischem Chrom(III)sulfat zudem Ameisensäure als Komplexbildner und Natriumsulfat als Leitsalz. Als Puffer werden Borsäure und Aluminiumsulfat eingesetzt. Zudem werden dem Elektrolyten organische Additive in Form von Natriumlaurylsulfat und Saccharin zugegeben. Das Verhältnis der Konzentrationen an Metallionen zur Konzentration an Komplexbildner wird bei allen Versuchen konstant bei 1:1,5 gehalten. Der pH-Wert des Elektrolyten beträgt 2,0 bis 2,1 bei einer Beschichtungstemperatur von 30 °C bis 50 °C.


Abb. 1: Beschichtungsprobe (Stahl-Rundstab, Ø = 10 mm, l = 30 mm)
Für die Beschichtungen werden Rundstäbe (Werkstoff 1.0122) mit einem Durchmesser von 10 mm und einer Länge von 30 mm verwendet (Abb. 1). Zur Einstellung einer definierten und gleichmäßigen Oberflächenbeschaffenheit werden die Rundstäbe mit Siliziumcarbid-Schleifpapier mit 500er- und 1000er-Körnung geschliffen. Um eine ausreichende Schichthaftung zu gewährleisten, werden die Proben vor der Beschichtung elektrolytisch entfettet (HSO UNI I, Herbert Schmidt GmbH) und in 10 Vol.-%iger Schwefelsäure aktiviert.
Die Beschichtungsversuche werden in einem Becherglas mit zwei parallel angeordneten Anoden durchgeführt (Abb. 2). Der Elektrolyt wird mittels einer Rührheizplatte temperiert. Der Rundstab wird über ein Labor-Rührwerk mit einer Umdrehungsgeschwindigkeit von 60 U/min in Rotation versetzt. Die elektrische Kontaktierung erfolgt über einen Kohle-Schleifkontakt. Um die Bildung von Chrom(VI) zu verhindern, werden Anoden mit einer Iridium-Mischoxidbeschichtung eingesetzt.

Abb. 2: Aufbau für die Beschichtungsversuche
4 Ergebnisse und Diskussion
In den Vorversuchen wurden unterschiedliche Elektrolytsysteme untersucht und grundlegende Beschichtungsparameter festgelegt. Die Beschichtung erfolgte anfangs noch auf Stahl- beziehungsweise Messingblechen. Es zeigte sich schnell, dass die Stromdichteverteilung und die Hydrodynamik im Elektrolyten großen Einfluss auf die Legierungszusammensetzung der abgeschiedenen Schicht nehmen. Da die Elektrolytbewegung mit einem Magnetrührstäbchen erfolgte, konnte keine gleichmäßige Anströmung der Kathode sichergestellt werden. Des Weiteren lag keine optimale primäre Stromdichteverteilung vor. An den Kanten des Blechs kam es zu einer Konzentration der elektrischen Feldlinien und somit zu einer ungleichmäßigen Stromdichteverteilung auf dem gesamten Blech.
Aufgrund dieser Problematik wurde der Versuchsaufbau angepasst. Durch Rotation eines Rundstabs sollte eine gleichmäßigere Hydrodynamik und Stromdichteverteilung sichergestellt werden. Durch den neuen Versuchsaufbau (Abb. 2) konnte eine gleichmäßigere Beschichtung erzielt werden. Die primäre Stromdichteverteilung war jedoch weiterhin nicht optimal, wodurch an den Kanten der beschichteten Probe eine höhere Schichtdicke und eine abweichende Legierungszusammensetzung vorlagen (Abb. 3). Aufgrund der starken Stromdichteabhängigkeit der Legierungszusammensetzung ist der Chromgehalt an den Kanten erhöht.

Abb. 3: Schichtdickenverteilung und Legierungszusammensetzung entlang der Längsachse des beschichteten Rundstabs
Als Ursache für die festgestellte starke Abhängigkeit der Legierungszusammensetzung von der Stromdichte kommt die großen Differenz zwischen den Abscheidepotentialen von Nickel (E0 = -230 mV) und Chrom (E0 = -760 mV) in Betracht. Damit Nickel und Chrom simultan abgeschieden werden können, muss kathodisch stark polarisiert werden. Die angewendeten Stromdichten sind dementsprechend hoch. Um einen ausreichend hohen Chromgehalt in der Legierungsschicht zu erhalten, wird die partielle Stromdichte der Nickelabscheidung durch Diffusionslimitierung begrenzt; das heißt, die Abscheidung von Chrom erfolgt durchtrittskontrolliert, wohingegen die Abscheidung von Nickel diffusionskontrolliert erfolgt.
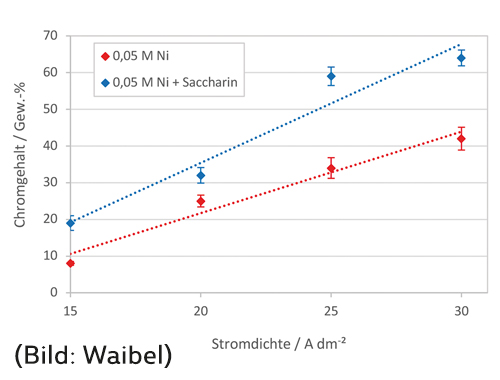
In Abbildung 4 ist der Einfluss der Stromdichte auf den Chromgehalt in der abgeschiedenen Schicht dargestellt. Durch Erhöhung der Stromdichte von 15 A/dm2 auf 30 A/dm2 steigt der Chromgehalt von 8 Gew.-% auf 42 Gew.-% an. Dies kann dadurch erklärt werden, dass durch Erhöhen der Gesamtstromdichte die Partialstromdichte der Chromabscheidung erhöht wird, wohingegen die Partialstromdichte der Nickelabscheidung aufgrund der Diffusionslimitierung konstant bleibt. Die Zugabe von Saccharin bewirkt zudem eine deutliche Erhöhung des Chromgehalts.

Abb. 4: Einfluss der Stromdichte auf den Chromgehalt (T = 40 °C)
Über die Wirkungsweise von Saccharin sind mehrere Mechanismen denkbar [5, 12]. Es ist davon auszugehen, dass Saccharin insbesondere die Chromreduktion beeinflusst und die parallel ablaufende Nickel- und Wasserstoffreduktion von sekundärer Bedeutung sind. Über das deprotonierte Stickstoffatom oder das carbonylische Sauerstoffatom kann Saccharin mit dem Metallion einen Komplex ausbilden [19]. Aufgrund der im Vergleich zur Metallionenkonzentration (bis zu 0,8 M) sehr geringen Saccharinkonzentration (0,008 M), ist eine Komplexierung von Chrom(III) oder Nickel(II) beziehungsweise eine selektive Komplexierung von Nickel(II) unwahrscheinlich. Die Reduktion von Chrom(III) ist ein mehrstufiger Prozess, der über die Zwischenstufe Chrom(II) abläuft. Die Reduktion von Chrom(II) zu Chrom(0) ist der geschwindigkeitsbestimmende Schritt in der gesamten Abscheidungsreaktion. Chrom(II) ist in wässriger Lösung jedoch nicht stabil und wird leicht zu Chrom(III) oxidiert. Um ein genügend hohes Angebot an Chrom(II) zu gewährleisten, muss dieses durch Komplexbildung stabilisiert werden. In der Annahme, dass die Kinetik der Ameisensäure nicht ausreichend schnell ist, um Chrom(II) zu stabilisieren, geschieht dies durch Saccharin. Da nur im kathodennahen Bereich eine Stabilisierung erfolgen muss, reichen bereits sehr geringe Mengen an Saccharin aus.
Die Erhöhung der Nickel(II)konzentration von 0,05 M auf 0,20 M führt dazu, dass der Chromgehalt in den abgeschiedenen Schichten abnimmt (Abb. 5). Zurückzuführen ist das auf die erhöhte Diffusionsgrenzstromdichte der Nickelabscheidung bei höheren Konzentrationen an Nickel(II).

Abb. 5: Einfluss der Nickelkonzentration auf den Chromgehalt (T = 40 °C, Elektrolyt enthält Saccharin)

Abb. 6: Einfluss der Peakstromdichte auf den Chromgehalt (T = 40 °C, ton = toff = 5 ms, Elektrolyt enthält Saccharin)
Neben der Abscheidung mit Gleichstrom wurde auch die Anwendung von pulsierendem Strom (Pulse Plating) untersucht. Für alle Beschichtungen wurde ein Duty Cycle von 50 % und eine Frequenz von 100 Hz gewählt, das heißt, die Pulszeit (ton) und die Pulspause (toff) betrugen jeweils 5 ms. Durch Steigerung der Peakstromdichte ip von 30 A/dm2 auf 50 A/dm2 konnte der Chromgehalt von 16 Gew.-% auf 55 Gew.-% erhöht werden (Abb. 6). Bei höheren Stromdichten war die Wasserstoffentwicklung so stark ausgeprägt, dass die Pufferkapazität nicht mehr ausreichte, um den pH-Wert im kathodennahen Raum zu stabilisieren. An der Kathodenoberfläche bildeten sich Hydroxyverbindungen, die eine weitere Abscheidung verhinderten. Im Vergleich zur Gleichstromabscheidung waren deutlich höhere Stromdichten erforderlich, um einen vergleichbaren Chromgehalt zu erzielen. In der Pulspause haben die Nickelionen die Möglichkeit, an die Kathode nach zu diffundieren. Dies führt zu einer höheren Nickelkonzentration an der Kathodenoberfläche wodurch die Diffusionsgrenzstromdichte der Nickelabscheidung erhöht wird.
Die Erhöhung der Beschichtungstemperatur von 30 °C auf 50 °C führt zu einer deutlichen Abnahme des Chromgehalts von 70 Gew.-% auf 16 Gew.-% (Abb. 7). Da die Diffusionsgeschwindigkeit mit zunehmender Temperatur steigt, erhöht sich die Diffusionsgrenzstromdichte der Nickelabscheidung. Die Chromabscheidung läuft hingegen durchtrittskontrolliert ab, das heißt, die Diffusion stellt nicht den geschwindigkeitsbestimmenden Schritt dar. Da eine Temperaturerhöhung zugleich depolarisierend wirkt, ist davon auszugehen, dass die Erhöhung der Diffusionsgeschwindigkeit und die Verringerung der Überspannung für Chrom(III) kleiner als für Nickel(II) ist, was schlussendlich den Chromgehalt in der Beschichtung verringert.

Abb. 7: Einfluss der Beschichtungstemperatur auf den Chromgehalt (ip = 40 A/dm2, ton = toff = 5 ms, Elektrolyt enthält Saccharin)
Die chemische Zusammensetzung der Schicht wurde mittels energiedispersiver Röntgenspektroskopie (EDX) ermittelt. Neben Chrom und Nickel weisen die abgeschiedenen Schichten nichtmetallische Einschlüsse auf. In allen Schichten wurden Sauerstoff und Kohlenstoff nachgewiesen. Der Kohlenstoffgehalt variiert dabei zwischen 0,7 Gew.-% und 3,0 Gew.-%. Insbesondere Ameisensäure, die im Elektrolyten als Komplexbildner enthalten ist, führt zu vergleichsweise hohen Kohlenstoffgehalten.
Der Einbau von Kohlenstoff bietet die Möglichkeit, durch eine anschließende Wärmebehandlung und der Ausbildung von Chromcarbiden, die Schichthärte zu erhöhen. Die Bildung von Chromcarbiden hat jedoch den Nachteil, dass der Anteil an freiem Chrom in der Schicht reduziert wird, was die Ausbildung einer Passivschicht erschweren kann. Wenn im Elektrolyt Saccharin enthalten ist, weisen die Schichten zudem geringe Mengen an Schwefel auf (0,3 Gew.-%–0,5 Gew.-%), was besonders im Hinblick auf eine Hochtemperaturanwendung kritisch zu sehen ist.
Die Morphologie der abgeschiedenen Schicht ist stark abhängig von der Oberflächengüte des Substrats. Aufnahmen mittels Rasterelektronenmikroskop (REM) lassen bei allen Schichten eine Vorzugsorientierung erkennen, die durch die Schleifriefen auf der Substratoberfläche verursacht wurde (Abb. 8–10). Das zeigt, dass der Elektrolyt eine geringe Mikrostreufähigkeit besitzt und die Oberflächenstruktur des Substrats nachgebildet wird. Nicht zu vernachlässigen ist zudem der Einfluss der Wasserstoffentwicklung. Aufgrund der geringen Stromausbeuten geht die Metallabscheidung mit einer starken Wasserstoffentwicklung einher. Der Wasserstoff entweicht dabei senkrecht beziehungsweise in Richtung der Schleifriefen nach oben, was den Effekt der vorwiegend auftretenden Orientierung verstärken kann. Die Schichten wachsen kompakt mit einer leicht globularen Struktur auf.

Abb. 8: REM-Aufnahme einer Nickel-Chrom-Schicht mit 22 Gew.-% Chrom (i = 20 A/dm2)

Abb. 9: REM-Aufnahme einer Nickel-Chrom-Schicht mit 31 Gew.-% Chrom (i = 25 A/dm2)

Abb. 10: REM-Aufnahme einer Nickel-Chrom-Schicht mit 55 Gew.-% Chrom (ip = 50 A/dm2, ton = toff = 5 ms)
Die abgeschiedenen Schichten weisen zudem hohe innere Spannungen auf, welche sich unter Rissbildung abbauen. Die Höhe der inneren Spannungen beziehungsweise der daraus resultierenden Risse ist besonders vom Chromgehalt der Schicht abhängig. Bei einem Chromgehalt von weniger als 22 Gew.-% sind nur vereinzelt Risse zu erkennen (Abb. 8). Mit zunehmendem Chromgehalt nimmt die Rissdichte jedoch stark zu. Bei einem Chromgehalt von 31 Gew.-% ist die gesamte Oberfläche von einem dichten Rissnetzwerk durchzogen (Abb. 9).
Ein höherer Chromgehalt ist gleichbedeutend mit einer höheren Stromdichte und einer stärkeren Wasserstoffentwicklung. Dadurch kann sich vermehrt Wasserstoff in das Gefüge einlagern. Zudem besteht die Gefahr, dass es aufgrund des erhöhten pH-Werts im kathodennahen Raum zur Bildung von Hydroxyverbindungen kommt, welche in die abgeschiedene Schicht miteingelagert werden. Bei einem höheren Chromgehalt ist zudem die Bildung von Chromhydrid und die damit einhergehende Volumenkontraktion stärker ausgeprägt. Alle genannten Faktoren führen zu einem verstärkten Auftreten von Rissen.
Durch die hohen inneren Spannungen lassen sich mittels Gleichstrom nur geringe Schichtdicken realisieren. Bei Schichtdicken größer 5 μm reißen die Schichten zum Teil auf und lösen sich vom Substrat ab. Eine vielversprechende Möglichkeit, die inneren Spannungen zu senken, bietet das Pulse Plating. Bei der Abscheidung mittels pulsierendem Strom konnten auch bei einem Chromgehalt von 55 Gew.-% noch rissfreie Schichten hergestellt werden (Abb. 10). Zudem konnten deutlich höhere Schichtdicken von bis zu 15 μm erreicht werden. In den Pulspausen kann der entstandene Wasserstoff aus dem Gefüge effundieren und die gebildeten Hydride zersetzen sich. Dadurch werden die inneren Spannungen der Schicht verringert und die Rissbildung wird unterdrückt. Ab einem Chromgehalt von 60 Gew.-% traten jedoch auch unter den gewählten Puls-Parametern wieder vereinzelt Risse auf.
Mit zunehmendem Chromgehalt gehen die Schichten von einer (teil)kristallinen in eine röntgenamorphe Kristallstruktur über (Abb. 11). Bei einem Chromgehalt von 16 Gew.-% sind im Röntgenbeugungsdiagramm mehrere Beugungspeaks zu erkennen, die sowohl der Nickel- als auch der Chromphase zugeordnet werden können [20]. Wenn der Chromgehalt auf 47 Gew.-% steigt ist nur noch im 2θ-Bereich zwischen 38° bis 46° ein breiter Peak zu erkennen, was darauf hinweist, dass die Schicht eine röntgenamorphe Kristallstruktur besitzt.

Abb. 11: Röntgenbeugungsdiagramme von Nickel-Chrom-Schichten mit unterschiedlichem Chromgehalt (Abscheidung mittels Pulsstrom, ton = toff = 5 ms, ip = 30 A/dm2 (16 Gew.-% Cr), 40 A/dm2 (27 Gew.-% Cr), 45 A/dm2 (47 Gew.-% Cr)
5 Zusammenfassung
In der vorliegenden Arbeit wurden Untersuchungen zur galvanischen Abscheidung von Nickel-Chrom-Legierungsschichten durchgeführt. Aufgrund der großen Differenz zwischen den Abscheidepotentialen von Nickel und Chrom zeigt sich eine starke Abhängigkeit der Legierungszusammensetzung von der Stromdichte. Um Schichten mit einer gleichmäßigen Legierungszusammensetzung abzuscheiden, ist die primäre Stromdichteverteilung von besonderer Bedeutung. Damit ein ausreichend hoher Chromgehalt in der Legierungsschicht erzielt werden kann, muss die partielle Stromdichte der Nickelabscheidung durch Diffusionslimitierung begrenzt werden. Durch Variation der Stromdichte und der Nickelkonzentration im Elektrolyten konnte der Chromgehalt in den abgeschiedenen Schichten nahezu beliebig eingestellt werden. Die Zugabe von Saccharin erhöhte den Chromgehalt und die Stromausbeute der Metallabscheidung.
Eine der größten Herausforderungen bei der Abscheidung von Nickel-Chrom-Schichten sind die hohen inneren Spannungen in den abgeschiedenen Schichten, welche sich unter Rissbildung abbauen. Die Abscheidung von größeren Schichtdicken mit einem hohem Chromgehalt war mit Gleichstrom nicht möglich. Die Anwendung von Pulse Plating zeigt jedoch großes Potential. Durch Auswahl geeigneter Parameter war es möglich, Schichten von mehr als 10 μm abzuscheiden, welche zudem rissfrei waren.
Literatur
[1] V. S. Protsenko, F. I. Danilov: Chromium electroplating from trivalent chromium baths as an environmentally friendly alternative to hazardous hexavalent chromium baths: comparative study on advantages and disadvantages; Clean Techn Environ Policy 16 (2014), S. 1201–1206
[2] A. Sheibani Aghdam, S. R. Allahkaram, S. Mahdavi: Corrosion and tribological behavior of Ni–Cr alloy coatings electrodeposited on low carbon steel in Cr (III)–Ni (II) bath; Surface and Coatings Technology 281 (2015), S. 144–149
[3] C. U. Chisholm: Electrodeposition of Nickel-Chromium Alloys from Aqueous Electrolytes: Review and Experimental Studies; Transactions of the IMF 46 (1968), S. 147–157
[4] Z. Zeng, Y. Zhang, W. Zhao, J. Zhang: Role of complexing ligands in trivalent chromium electrodeposition; Surface and Coatings Technology 205 (2011), S. 4771–4775
[5] J. Datta: Chrom(lll)-Bäder: Grundlagen für die Entwicklung, Eigenschaften, Abscheidungsmechanismus; Galvanotechnik 73 (1982), S. 106–112
[6] F. I. Danilov, V. S. Protsenko, V. O. Gordiienko: Electrode processes occurring during the electrodeposition of chromium-carbon coatings from solutions of
Cr(III) salts with carbamide and formic acid additions; Russ J Electrochem 49 (2013), S. 475–482
[7] F. I. Danilov, V. S. Protsenko, V. O. Gordiienko, A. S. Baskevich, V. V. Artemchuk: Electroplating of wear-
resistant nanocrystalline coatings from a bath containing basic chromium(III) sulfate (chrome tanning agent); Prot Met Phys Chem Surf 49 (2013), S. 299–303
[8] R. Giovanardi, G. Orlando: Chromium electrodeposition from Cr(III) aqueous solutions; Surface and Coatings Technology 205 (2011), S. 3947–3955
[9] S. Surviliene, A. Češūnienė, A. Selskis, R. Butkienė: Effect of Cr(III)+Ni(II) solution chemistry on electrodeposition of CrNi alloys from aqueous oxalate and glycine baths; Transactions of the IMF 91 (2013),
S. 24–31
[10] J.-M. Langlais: Beitrag zur galvanischen Abscheidung von Eisen-Chrom-Nickel-Legierungen; Dissertation, Aachen, 1984
[11] J.-P. Nepper, Untersuchungen an galvanisch abgeschiedenen Eisen-Chrom-Nickel-Schichten: Struktur, Zusammensetzung und Korrosionsverhalten. Dissertation, Aachen, 1987
[12] M. Leimbach, C. Tschaar, U. Schmidt, A. Bund, Electrochemical characterization of chromium deposition from trivalent solutions for decorative applications by EQCM and near-surface pH measurements, Electrochimica Acta 270 (2018) 104–109
[13] J. Bohnet, Entwicklung eines Verfahrens zur Abscheidung technischer Chromschichten aus einem Chrom(III)-Elektrolyt. Dissertation, Stuttgart, 2009
[14] J. Wijenberg, A. de Vooys, R. Kortlever, M. Koper, Oxidation reactions in chromium(III) formate electrolytes at platinum and at a catalytic mixed metal oxide coating of iridium oxide and tantalum oxide, Electrochimica Acta 213 (2016) 194–200
[15] C. an Huang, C.Y. Chen, C.C. Chen, T. Kelly, H.M. Lin, Microstructure analysis of a Cr–Ni multilayer pulse-electroplated in a bath containing trivalent chromium and divalent nickel ions, Surface and Coatings Technology 255 (2014) 153–157
[16] L. Xu, Z. Gong, J. Tang, Q. He, N. He, J. Du, Ni-Cr alloy electrodepositing technology on Fe substrate and coating performance, J Cent. South Univ. Technol. 14 (2007) 181–185
[17] S. Kölle, Entwicklung eines hochbeständigen, galvanisch abgeschiedenen Schichtsystems für den Einsatz in maritimer Technik bei starker tribokorrosiver Belastung. Dissertation, Stuttgart, 2018
[18] K.-L. Lin, C.-J. Hsu, I.-M. Hsu, J.-T. Chang, Electroplating of Ni-Cr on steel with pulse plating, JMEP 1 (1992) 359–361
[19] E.J. Baran, V.T. Yilmaz, Metal complexes of saccharin, Coordination Chemistry Reviews 250 (2006) 1980–1999
[20] J. Sun, D.X. Du, H.F. Lv, L. Zhou, Y.G. Wang, C.G. Qi, Microstructure and corrosion resistance of pulse electrodeposited Ni–Cr coatings, Surface Engineering 31 (2015) 406–411
